
Controllable synthesis of Co1−x MxFe2O4 nanoparticles (M = Zn, Cu, and Mn; x = 0.0 and 0.5) by cost-effective sol–gel approach: analysis of structure, elastic, thermal, and magnetic properties
Substitutions of cations were considered to be the main way for improving the performance of ferrite nanocrystalline structures. In this paper, non-magnetic and magnetic ions were conducted to substitute cobalt spinel ferrite nanoparticles CoFe2O4 NPs (CFO NPs). The studied Co1−xMxFe2O4; M = Zn, Cu, and Mn; x = 0.00, and 0.50) samples were synthesized through a cost-effective sol–gel technique. The outstanding properties of the samples are addressed using XRD, FTIR, the inductively coupled plasma optical emission spectrometer (ICP-OES), Raman analyses, HR-TEM, BET surface area analyzer, the

Fabrication of Ultra-Pure Anisotropic Zinc Oxide Nanoparticles via Simple and Cost-Effective Route: Implications for UTI and EAC Medications
The purposes of this work are to evaluate the antimicrobial, antibiofilm, anticancer, and antioxidant abilities of anisotropic zinc oxide nanoparticles (ZnO NPs) synthesized by a cost-effective and eco-friendly sol–gel method. The synthesized ZnO NPs were entirely characterized by UV-Vis, XRD, FTIR, HRTEM, zeta potential, SEM mapping, BET surface analyzer, and EDX elemental analysis. Antimicrobial and antibiofilm activities of ZnO NPs were investigated against multidrug-resistant (MDR) bacteria and yeast causing serious diseases like urinary tract infection (UTI). The anticancer activity was
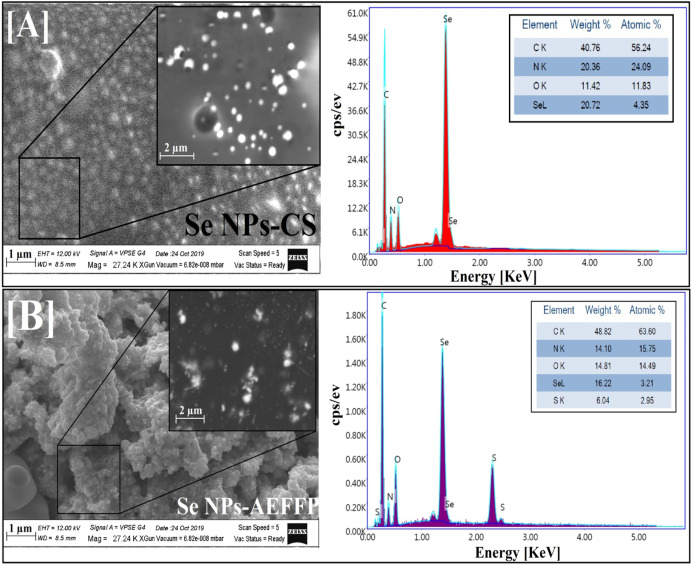
Factorial design-optimized and gamma irradiation-assisted fabrication of selenium nanoparticles by chitosan and Pleurotus ostreatus fermented fenugreek for a vigorous in vitro effect against carcinoma cells
The novelty of the present work looks in the synthesis of aqueous dispersed selenium nanoparticles (Se NPs) using gamma rays with the aid of various natural macromolecules such as citrus pectin (CP), sodium alginate (Alg), chitosan (CS) and aqueous extract of fermented fenugreek powder (AEFFP) using Pleurotus ostreatus for investigating their impact in vitro toward carcinoma cell. The synthesized Se NPs were characterized by XRD, UV–Vis., DLS, HRTEM, SEM, EDX and FTIR. Nucleation and growth mechanisms were also discussed. The factorial design was applied to examine the importance of multiple
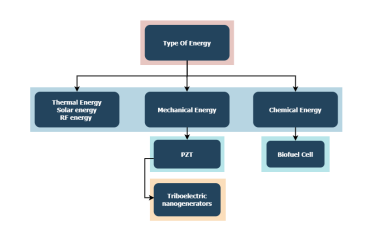
Study of Energy Harvesters for Wearable Devices
Energy harvesting was and still an important point of research. Batteries have been utilized for a long time, but they are now not compatible with the downsizing of technology. Also, their need to be recharged and changed periodically is not very desirable, therefore over the years energy harvesting from the environment and the human body have been investigated. Three energy harvesting methods which are the Piezoelectric energy harvesters, the Enzymatic Biofuel cells, and Triboelectric nanogenerators (TENGs) are being discussed in the paper. Although Biofuel cells have been investigated for a
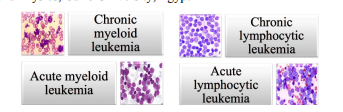
Fractional-order mathematical model for Chronic Myeloid Leukaemia
This paper is dedicated to develop a fractional order model of the rate of change of cancerous blood cells in Chronic Myeloid Leukaemia using fractional-order differential equations as well as tackling the factors that affect this rate and compare between them. The simulated cases (using MATLAB) prove that the proposed model is doable in terms of the variables positions in the equations and its effect on the overall population. Also, the effect of the Pactional order is investigated through three parameters sets and it has shown strong influence on the dynamic response. © 2017 IEEE.
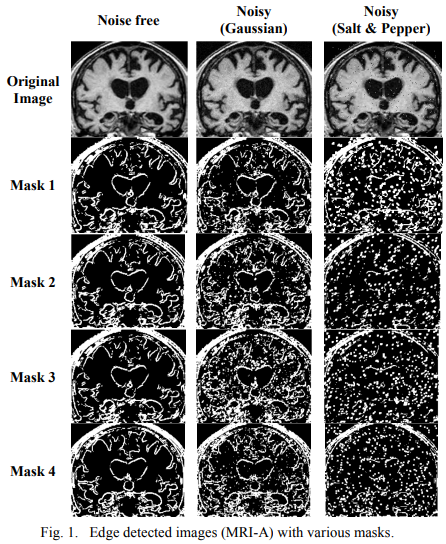
Comparative study of fractional filters for Alzheimer disease detection on MRI images
This paper presents a comparative study of four fractional order filters used for edge detection. The noise performance of these filters is analyzed upon the addition of random Gaussian noise, as well as the addition of salt and pepper noise. The peak signal to noise ratio (PSNR) of the detected images is numerically compared. The mean square error (MSE) of the detected images as well as the execution time are also adopted as evaluation methods for comparison. The visual comparison of the filters capability in medical image edge detection is presented, that can help in the diagnosis of
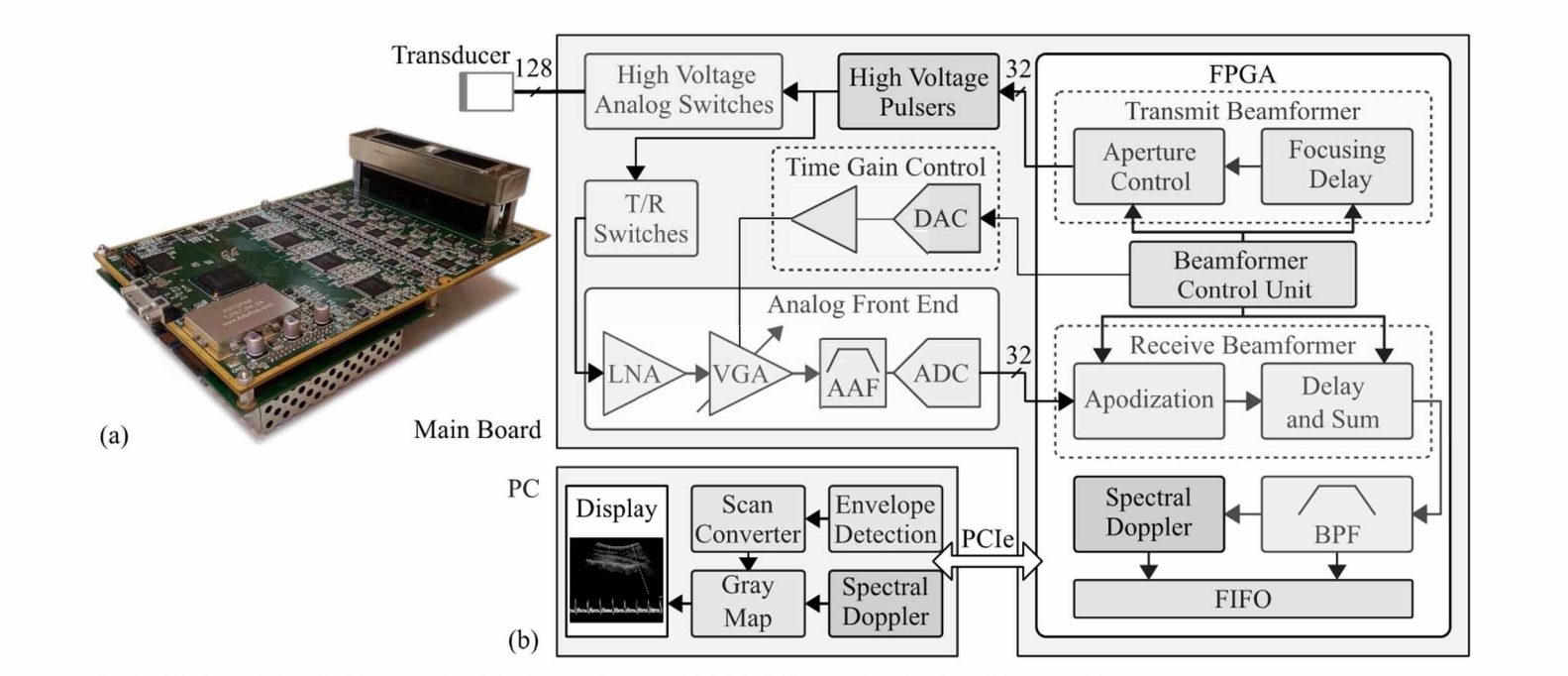
Implementation of a Pulsed-Wave Spectral Doppler Module on a Programmable Ultrasound System
Pulsed wave Doppler ultrasound is commonly used in the diagnosis of cardiovascular and blood flow abnormalities. Doppler techniques have gained clinical significance due to its safety, real-time performance and affordability. This work presents the development of a pulsed wave spectral Doppler module, which was integrated into a reconfigurable ultrasound system. The targeted system adopts a hardware-software partitioning scheme where an FPGA handles the front-end and a PC performs the back-end. Two factors were considered during the design. First, the data transfer rate between hardware and
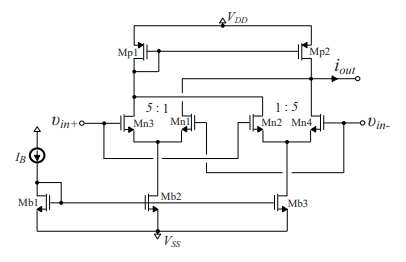
Design of fractional-order differentiator-lowpass filters for extracting the R peaks in ECG signals
An implementation of a fractional-order differentiator-lowpass filter is presented in this work, which is constructed from Operational Transconductance Amplifiers as active cells. This offers the benefits of electronic tuning and, also, of monolithic implementation. The presented scheme has been employed for the extraction of the R peaks in electrocardiogram signals due to its efficiency for performing this task even in a noisy environment. The provided post-layout simulation results confirm the correct operation of this solution as well as its reasonable sensitivity characteristics. © 2019
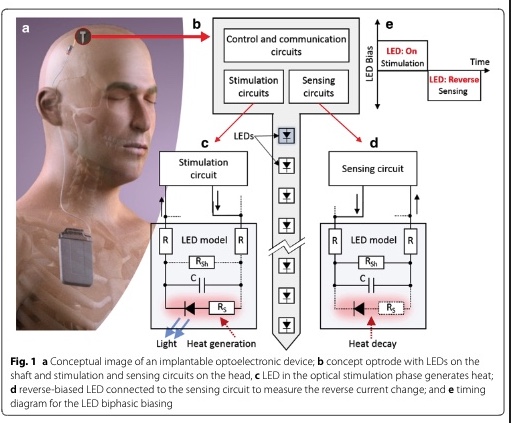
A current-mode system to self-measure temperature on implantable optoelectronics
Background: One of the major concerns in implantable optoelectronics is the heat generated by emitters such as light emitting diodes (LEDs). Such devices typically produce more heat than light, whereas medical regulations state that the surface temperature change of medical implants must stay below + 2 °C. The LED's reverse current can be employed as a temperature-sensitive parameter to measure the temperature change at the implant's surface, and thus, monitor temperature rises. The main challenge in this approach is to bias the LED with a robust voltage since the reverse current is strongly
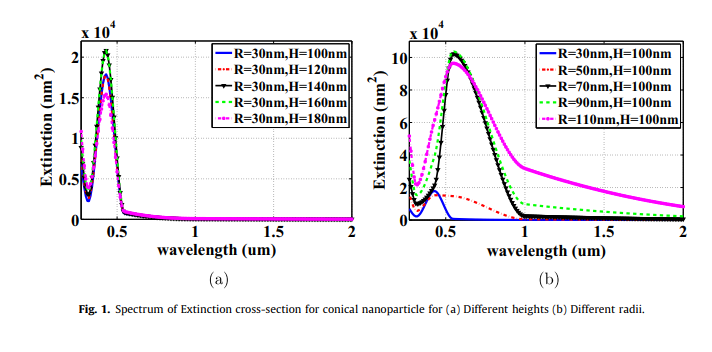
J-V characteristics of plasmonic photovoltaics with embedded conical and cylindrical metallic nanoparticles
Plasmonic photovoltaics (PVs) are promising structures that improve thin-film photovoltaics performance, where optical absorption is improved via embedding metallic nanoparticles in the PV's active layer to trap the incident optical wave into the photovoltaic cell. The presented work investigates the design of PV with both structures of conical and cylindrical metallic nanoparticles through studying their extinction cross-sections and electric field distributions. Also, the impact of these nanoparticles in silicon PVs on the optical absorption enhancement is investigated. The figure of merit